
里 INvestment:''和''而不同:一样/不一样的CSF1R(下篇)

已上市治疗cGVHD的药物
(1)Ibrutinib/艾伯维
伊布替尼在2017年获FDA批准治疗cGVHD,用于≥1L治疗,当时是该疾病领域第一个的获批药物。随后2022年,FDA也批准了1-12岁儿童低剂量使用的该项适应症。
“The U.S. Food and Drug Administration today expanded the approval of Imbruvica (ibrutinib) for the treatment of adult patients with chronic graft versus host disease (cGVHD) after failure of one or more treatments. This is the first FDA-approved therapy for the treatment of cGVHD.”
https://www.fda.gov/drugs/resources-information-approved-drugs/fda-expands-ibrutinib-indications-chronic-gvhd
--批准基于研究PCYC-1129-CA(NCT02195869),这是一项开放标签、多中心、单臂临床试验,纳入了42名在一线皮质类固醇治疗失败后需要额外治疗的慢性移植物抗宿主病(cGVHD)患者。大多数患者(88%)在基线时至少有两个器官受累。最常见的受累器官是口腔(86%)、皮肤(81%)和胃肠道(33%)。
--患者每日口服420 mg伊布替尼。研究者评估的ORR是67%,即28名患者(95% CI:51%,80%)。中位缓解时间(mDOR)为12.3周(即约3个月)(范围:4.1至42.1周)。在所有受累器官(皮肤、口腔、胃肠道和肝脏)中均观察到缓解。48%的患者(n=20)观察到持续五个月或更长时间的缓解。
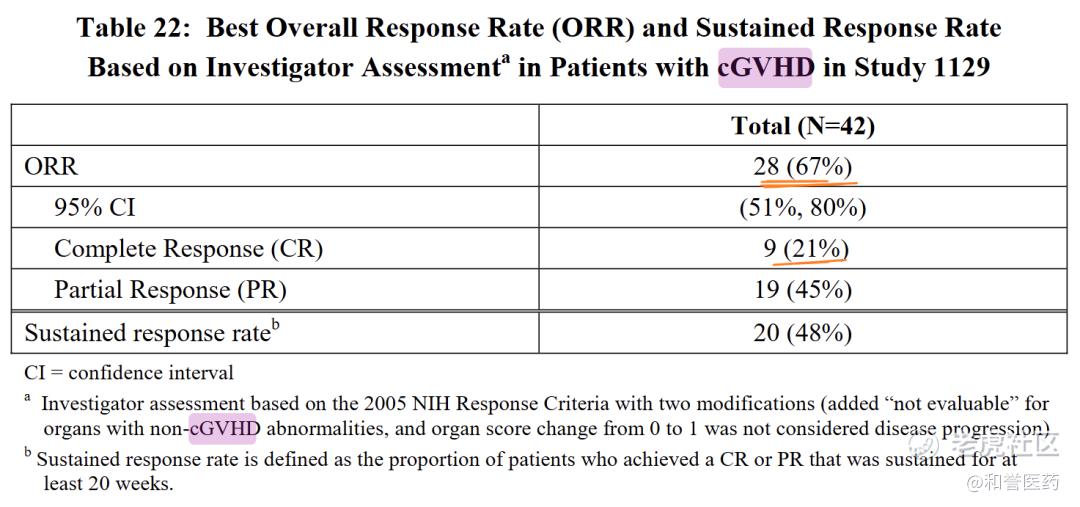
--推荐剂量为每日一次口服420毫克(即每日一次服用三粒140毫克胶囊)。

(2)Ruxolitinib/Incyte、Novartis
2009年,Incyte与诺华(Novartis)达成协议,将美国以外市场的商业化权利授予后者,商品名:Jakavi;而Incyte则负责芦可替尼在美国的开发和商业化,商品名为Jakafi。
贵为重磅的JAK抑制剂,芦可替尼适应症包括:中度或高危骨髓纤维化、成人真性红细胞增多症,除此之外,乳膏剂型(商品名为Opzelura)适应症还包括:12岁及以上轻中度特应性皮炎、成人和12岁及以上儿童患者的非节段性白癜风。该药于2011年11月首次在美国获批,2023年全球销售额突破43亿美元,跻身重磅药物行列。
当然,今天本文主要还是用来介绍cGVHD适应症的数据:
--2021年9月22日,美国食品药品监督管理局(FDA)批准Jakafi(中文:芦可替尼)用于治疗12岁及以上成人和儿童患者在一线或二线全身治疗失败后的慢性移植物抗宿主病(cGVHD)。
--疗效评估基于REACH-3研究(NCT03112603),这是一项随机、开放标签、多中心的临床试验,比较了芦可替尼与最佳可用疗法(BAT)在异基因干细胞移植后对皮质类固醇难治性cGVHD的治疗效果。试验将329名患者随机(1:1)分配接受芦可替尼10 mg每日两次或BAT治疗。
--支持批准的主要疗效终点是第7周期第1天的总体反应率(ORR)(2014年NIH反应标准)。芦可替尼组的ORR为70%(95% CI 63%,77%),BAT组为57%(95% CI 49%,65%),反应率差异为13%(95% CI 3%,23%)。从首次反应到疾病进展、死亡或开始新的慢性GVHD全身治疗的中位反应持续时间mDOR,芦可替尼组为4.2个月(95% CI 3.2,6.7),BAT组为2.1个月(95% CI 1.6,3.2)。从首次响应到死亡或开始新的cGVHD全身治疗的中位时间,芦可替尼组为25个月(95% CI 16.8,NE),BAT组为5.6个月(95% CI 4.1,7.8)。
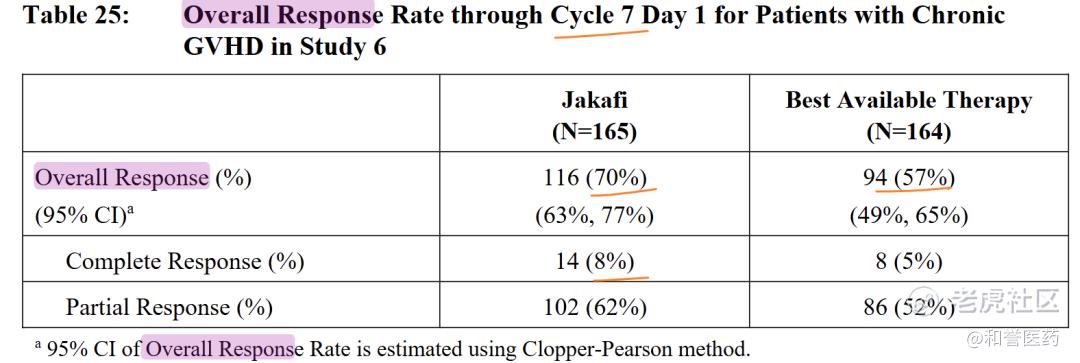
(Cycle 7 Day 1 (each cycle was comprised of 4 weeks))
--在cGVHD患者中,芦可替尼最常见的(发生率>35%)血液学不良反应是贫血和血小板减少。最常见的(发生率≥20%)非血液学不良反应是感染和病毒感染。
--芦可替尼用于cGVHD的推荐起始剂量为10 mg,每日两次口服。
On September 22, 2021, the Food and Drug Administration approved ruxolitinib (Jakafi, Incyte Corp.) for chronic graft-versus-host disease (cGVHD) after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older.
https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-chronic-graft-versus-host-disease
(3)Axatilimab/Incyte
2024年8月14日,美国食品药品监督管理局(FDA)批准Axatilimab(商品名:Niktimvo)上市,用于治疗至少接受过两种系统性治疗失败的慢性移植物抗宿主病(cGVHD)成人和体重至少40公斤的儿童患者。Axatilimab由美国生物制药公司Incyte研发(没错,~~Incyte again)。
--Axatilimab的疗效在AGAVE-201研究(NCT04710576)中进行了评估。AGAVE-201是一项随机、开放标签、多中心的2期临床试验,主要研究了3种剂量的Axatilimab在复发或难治性cGVHD成人和儿童患者中的疗效。这些患者此前已接受过至少两种系统性治疗,并且需要额外治疗。
--Axatilimab的给药方式是静脉输注。对于体重至少40公斤的患者,推荐的剂量为0.3 mg/kg,最大剂量为3mg,每次静脉输注30分钟,每2周一次,直到疾病进展或出现无法耐受的毒性。
--在接受推荐剂量的Axatilimab治疗的79名患者中:客观缓解率(ORR)为75%;获得客观缓解的患者平均在治疗1.5个月时,疾病开始出现缓解。
--患者中位缓解时间mDOR为1.9个月。在获得缓解的患者中,有60%的患者疾病至少稳定了12个月,疾病没有恶化、出现死亡或需要更换治疗方案。
--Axatilimab治疗最常见的不良反应包括天冬氨酸转氨酶(AST)升高、感染(病原体不明)、丙氨酸转氨酶(ALT)升高、磷酸盐降低、血红蛋白降低、病毒感染、γ-谷氨酰转移酶(GGT)升高、肌肉骨骼疼痛、脂肪酶升高、疲劳、淀粉酶升高、钙升高、肌酸磷酸激酶(CPK)升高、碱性磷酸酶(ALP)升高、恶心、头痛、腹泻、咳嗽、细菌感染、发热和呼吸困难。不良反应的发生率和分级具有Axatilimab剂量依赖性。49%的0.3mg剂量组患者,60%的1mg剂量组和71%的3mg剂量组的患者发生3级及以上的不良反应。
https://mp.weixin.qq.com/s/TEKn0gsyt1bauCAXM1aAQQ
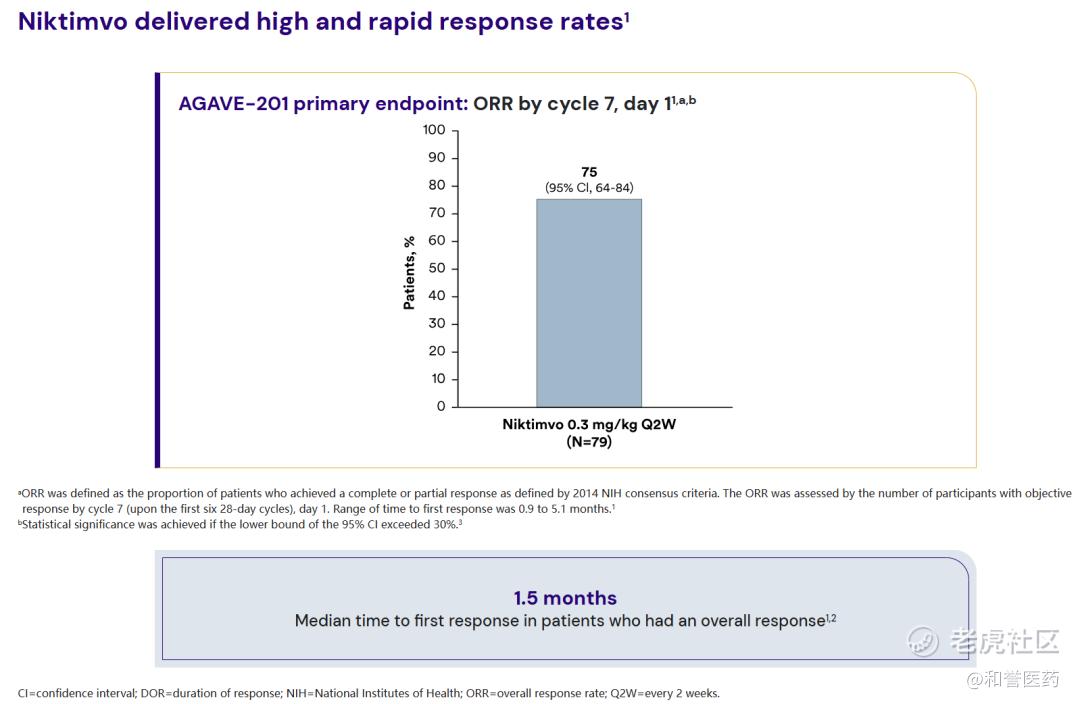
*另外两个剂量组1.0mg和3.0mg的ORR也达到67%和50%。中位反应时间为1.5个月。
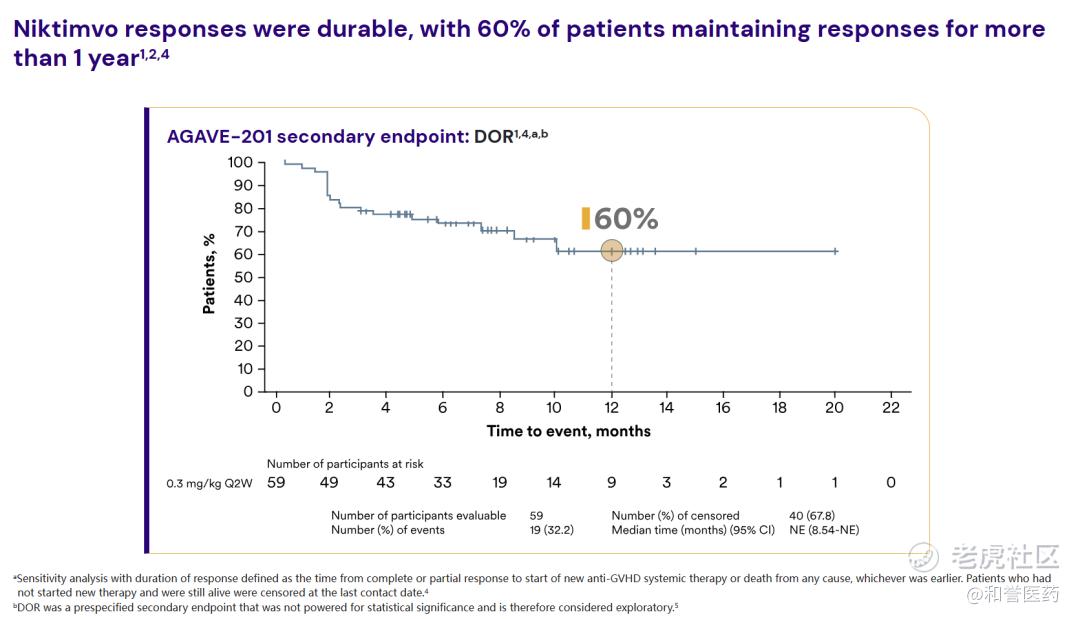
*在获得缓解的患者中,有60%的患者疾病至少稳定了12个月。
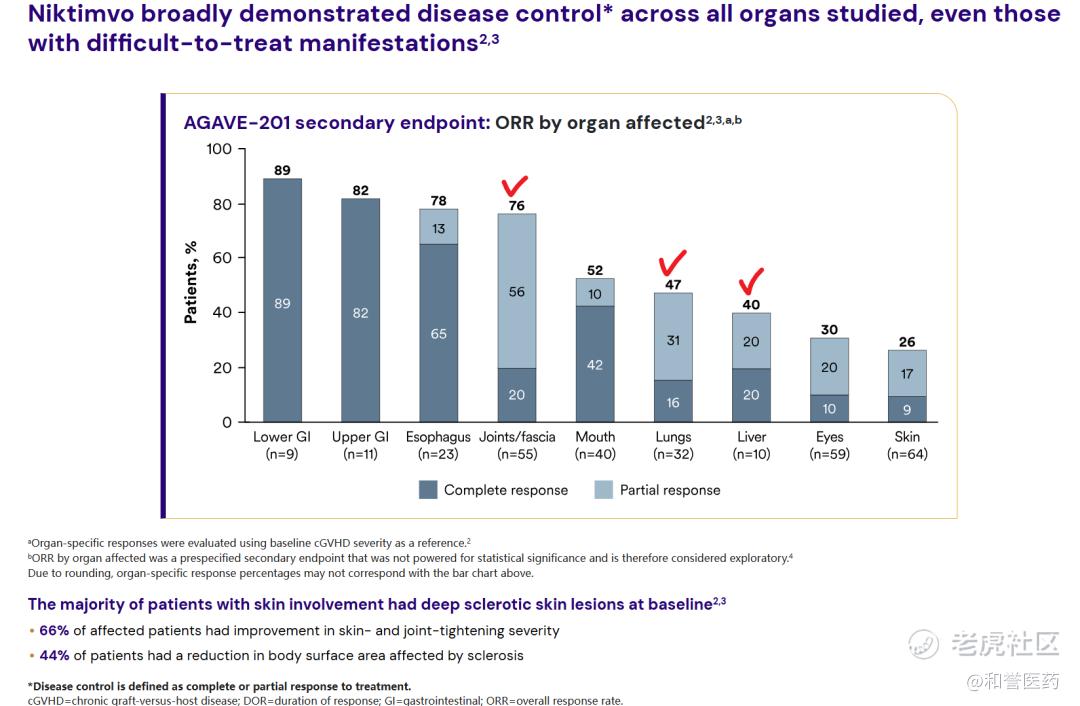
*不同部位/器官的ORR数据,关节/筋膜:76%(CR:20%)、肺部:47%(CR:16%)、肝脏:40%(CR:20%)。
(4)Belumosudil/Sanofi
Rezurock® (Belumosudil)(中文:贝舒地尔) is the first and only approved therapy inhibiting Rho-associated coiled-coil kinase 2 (ROCK2). Rezurock is approved in the United States for the treatment of adult and pediatric patients 12 years and older with cGVHD after failure of at least two prior lines of systemic therapy. (不翻了,累了)
--贝舒地尔在2021年获FDA批准上市,2023年获中国CDE批准上市,另外英国、加拿大和澳大利亚也已经获批上市,目前治疗范围是3L疗法获批,同时1L疗法已经进入到ph3阶段。(作者认为,cGVHD一旦无效就可以换药,所以这里的三线治疗不是严格意义上的三线。)
--2021年9月8日,跨国制药巨头赛诺菲宣布与Kadmon Holdings签订最终收购协议,每股9.50美元现金收购Kadmon全部股份,较9月7日收盘价每股5.3美元溢价79.2%,收购金额总计约19亿美元,Kadmon的核心资产为Belumosudil/KD025。(在收购前该药已经获得FDA批准上市)
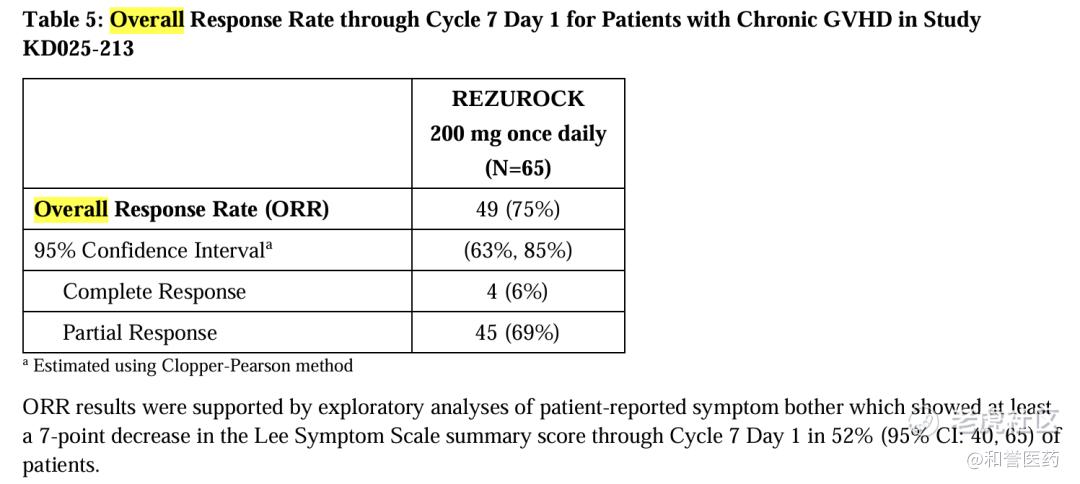
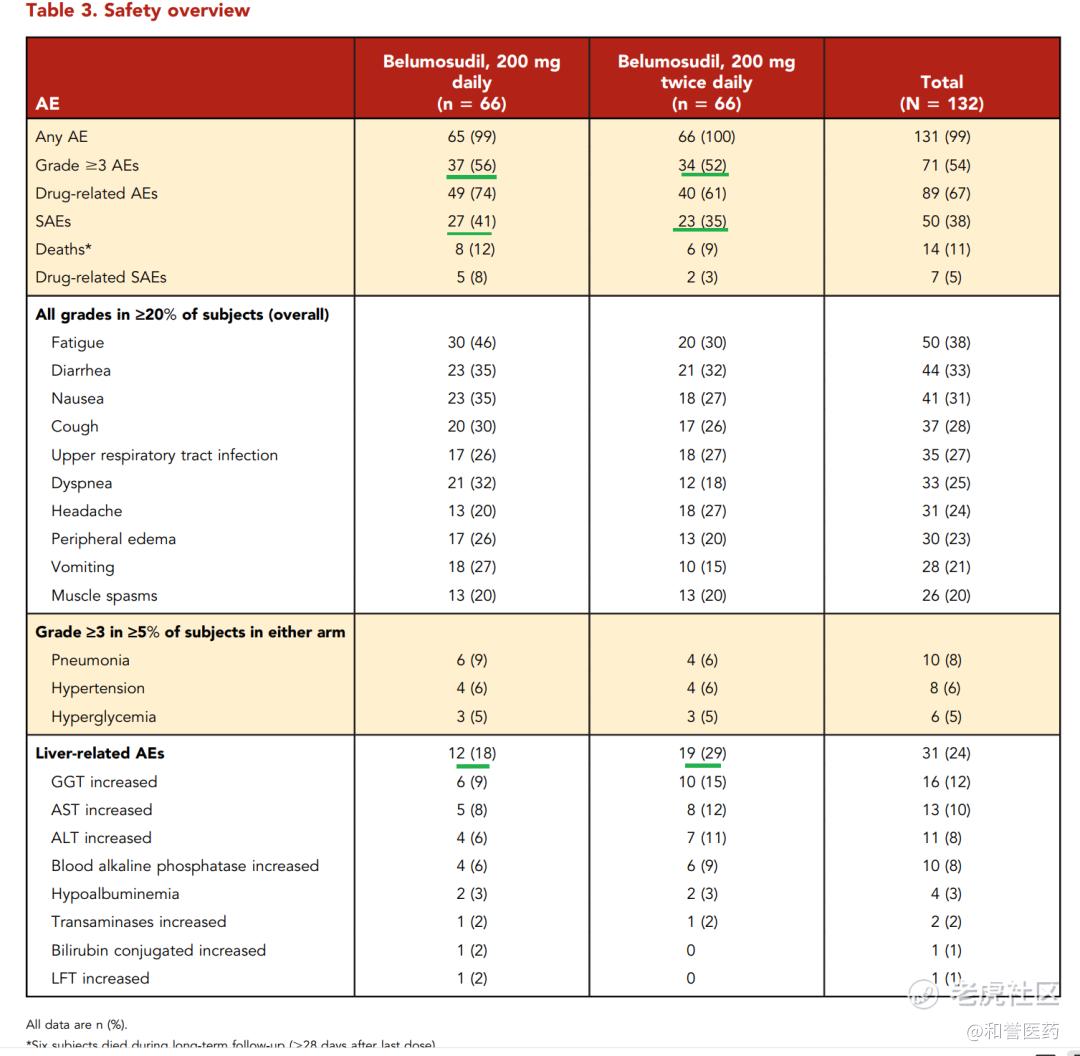
--在ROCKstar/NCT03640481、KD025-213(随机、开放标签、多中心关键性)临床试验临床中,共计有65名患者接受了belumosudil的治疗。试验结果显示:belumosudil治疗组患者的总缓解率(ORR)为75%,完全缓解率为6%;中位缓解持续时间mDOR为1.9个月,出现首次缓解所需的中位时间为1.8个月,62%获得缓解的患者在缓解后至少12个月不需要使用新的全身性疗法。
--ROCKstar 设计了两个队列,都是分别是200mgQD和200mgBID,结果发现每日一次和每日两次的疗效一样,但每日一次的安全性更好,因此最后只是根据QD的队列进行上市申请。
--Belumosudil的相对耐受性良好,中位相对剂量强度(RDI)为99.7%。81%的受试者的RDI>95%。接受CS和其他免疫抑制剂的cGVHD患者的AEs与预期一致。38%的受试者发生了≥1种SAE;最常见的是肺炎(7%)。最常见的(≥5%)3级或4级AE是肺炎(8%)、高血压(6%)和高血糖(5%)。最常见的肝脏相关AE是谷丙转氨酶升高(12%)。
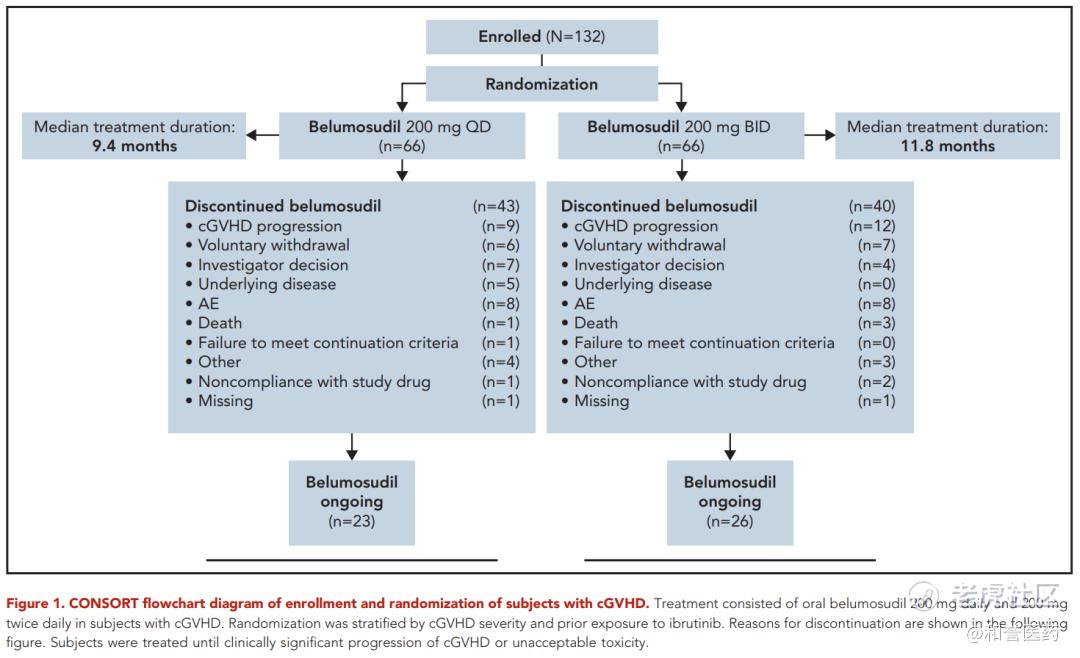
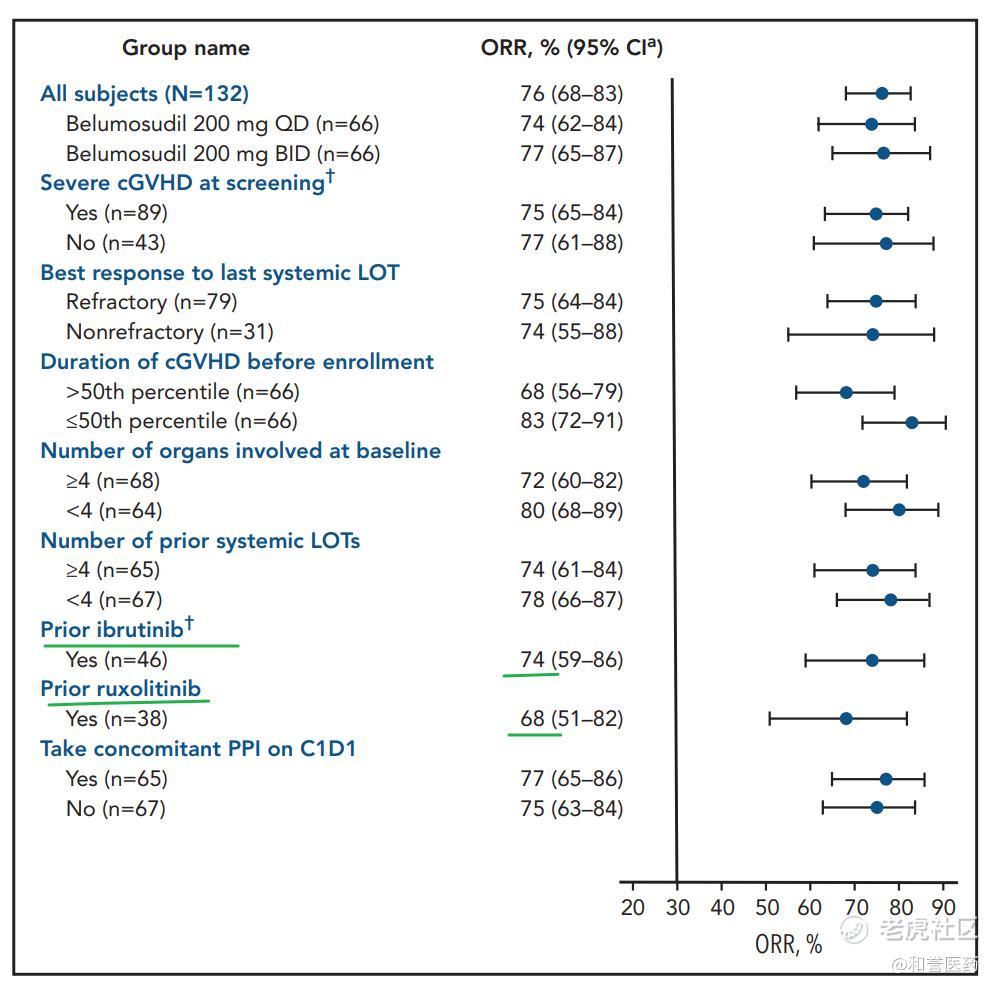
*可以看到在伊布替尼芦可替尼经治患者中,仍然可以获得74%和68%的ORR效果,此药获批一线疗法基本没有问题。
Corey Cutler, Stephanie J. Lee, Sally Arai,Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the ROCKstar Study
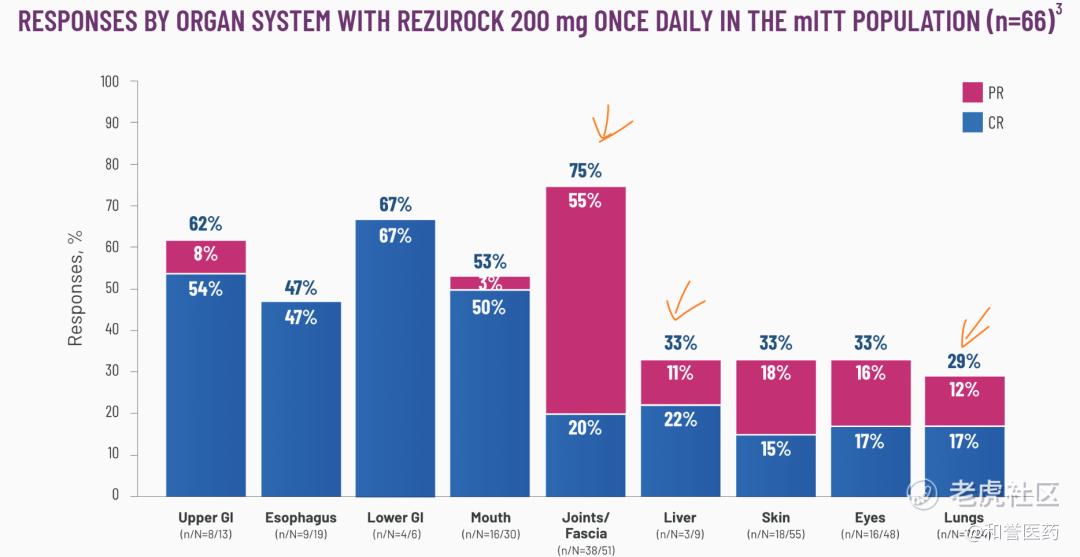
*不同部位/器官的ORR数据,关节/筋膜:75%(CR:20%)、肺部:29%(CR:17%)、肝脏:33%(CR:22%),数据稍逊于Incyte的CSF1R单抗。
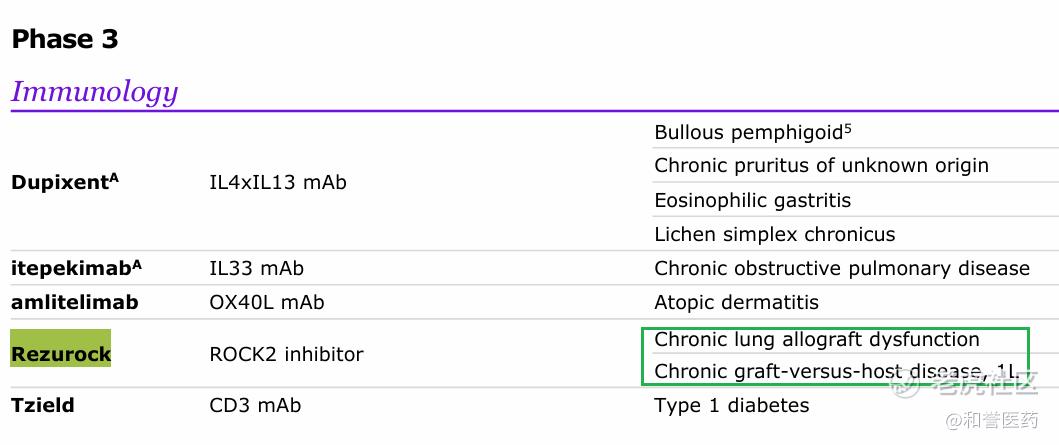
*除了一线疗法的临床之外,贝舒地尔还开始针对CLAD开发专门针对肺移植的适应症。根据国际心肺移植协会(ISHLT)的数据报道,目前全球每年有大于四千多名患者接受肺移植,迄今为止,全球完成的肺移植总例数大于10万。但是,和其它实体器官移植相比,肺移植术后的长期存活率仍不理想。ISHLT的数据显示:目前肺移植术后全球的平均中位生存期仅6.5年。影响肺移植术后远期存活的原因是各种并发症,其中,慢性移植肺失功(chronic lung alograft dysfunction, CLAD)是肺移植术后远期的主要并发症,也是影响远期存活的首要并发症。实际上,肺移植术后并发的CLAD和HSCT后肺部的cGVHD,二者的发病机理非常相似,均属于免疫反应介导的慢性炎症反应性导致的小气道-肺疾病。(目前正在进行的ph3全球多中心临床:ROCKaspire)
https://mp.weixin.qq.com/s/5qX9arcD_MZNf6nhSdOAtw
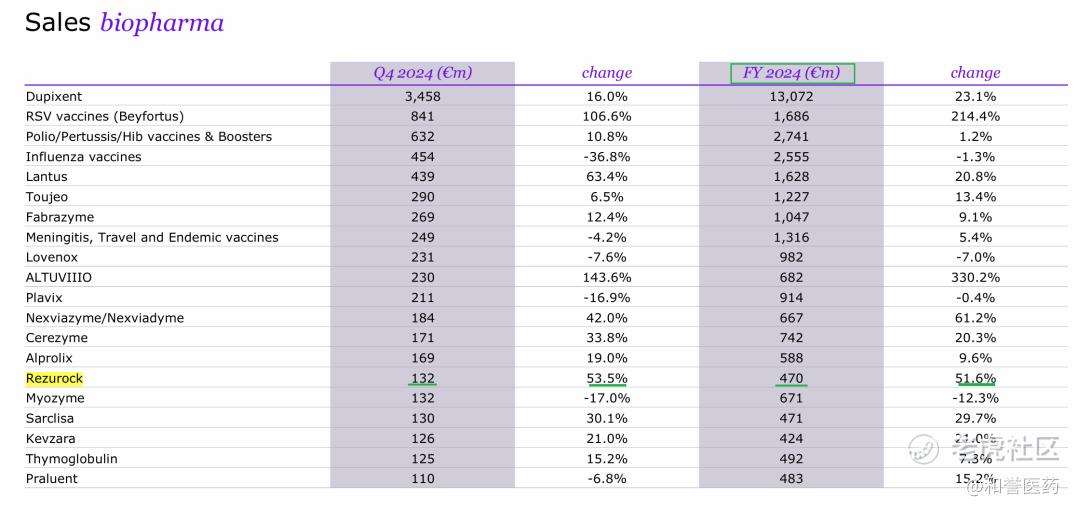
*2024年卖了4.7E欧,还是在yoy+51.6%,上市三年还有这个增速,后面还有1线cGVHD和CLAD的适应症在ph3,预计峰值还会继续拔高,1billion好像也不是问题了。

研发中的cGVHD药物:JAK/ROCK2抑制剂/正大天晴
Janus激酶(JAK)信号通路在免疫细胞活化和组织炎症中起着重要作用,ROCK2信号通路则在通过上调STAT3则在多种纤维化途径中发挥核心作用,从而促进慢性GVHD的进展。罗伐昔替尼(TQ05105,正大天晴自主研发)是一种新型的、口服的双重JAK1/2和ROCK1/2抑制剂,作用机制针对慢性GVHD的炎症和纤维化成分,或将为治疗慢性GVHD提供了一种新方法。
以下是关于临床结果的数据:
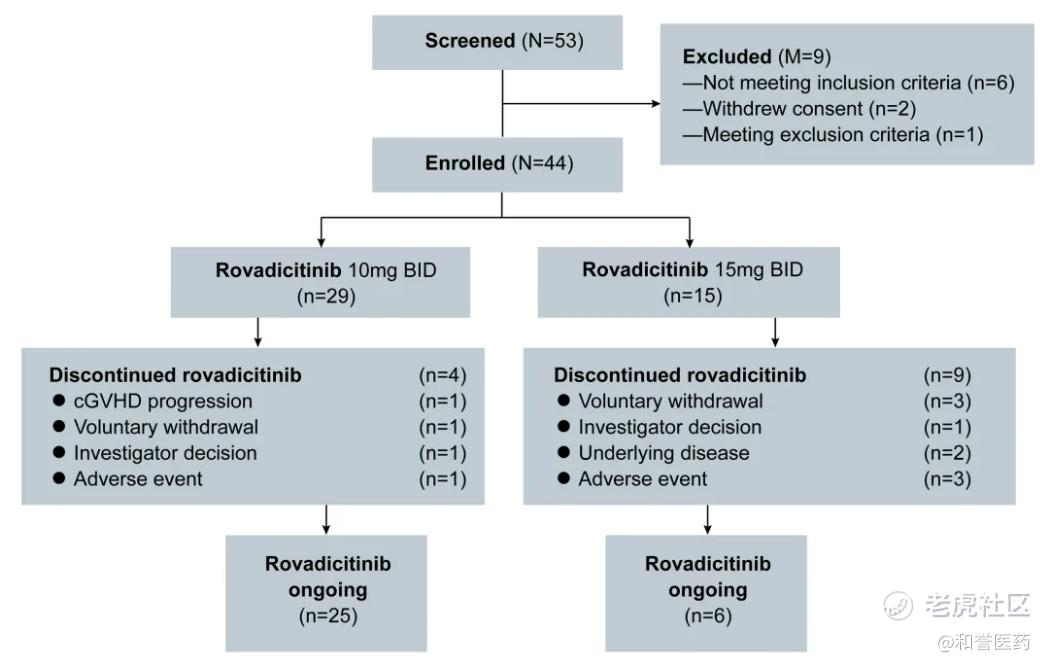
--这项多中心、开放标签、1b/2a期临床试验(NCT04944043)在中国的7个移植中心进行,招募了中度或重度激素难治性或依赖性慢性GVHD患者。研究采用3+3设计,分为Ib和II期研究两个部分。1b期剂量探索使用了标准的3+3设计,探索两个剂量(罗伐西替尼10mg BID和15mg BID)的安全性,II期为扩充队列研究,进一步明确RP2D及药物有效性。患者口服罗伐昔替尼至少6个周期(每个周期28天),直到慢性GVHD进展或出现不可接受的毒性。
--研究共筛选了53例患者中,有44名患者入组并纳入分析。患者中位年龄为34岁(范围,16-59岁),其中27%(61.4%)患者为男性。根据2014年NIH共识标准,入组患者的慢性GVHD严重程度占比为中度36.4%和重度63.6%。29名(65.9%)患者症状累及≥4个器官,26名(59.1%)有肺受累者,其中23.1%的患者NIH肺评分为3分。36例(81.8%)患者接受了≥2线系统性治疗,其中,18例(40.9%)既往接受芦可替尼治疗,14例患者对芦可替尼耐药。
疗效方面:
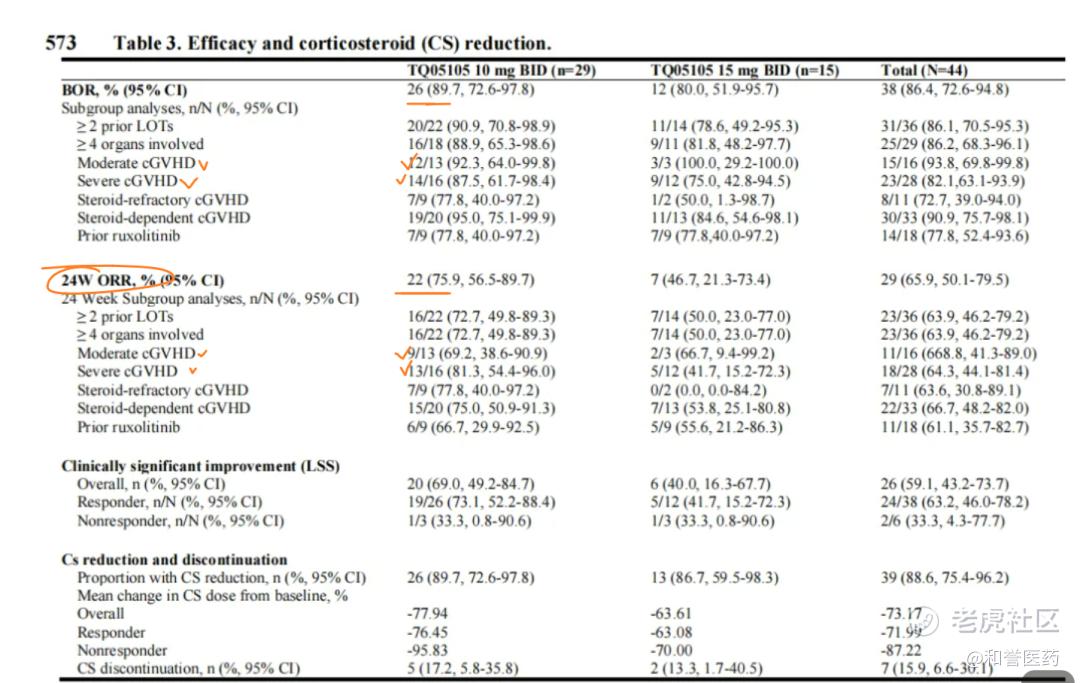
--罗伐昔替尼对既往使用了芦可替尼的患者仍然有效,缓解率为77.8%。
--治疗数据:罗伐昔替尼治疗最佳缓解率(BOR)为86.4%,10mg BID队列最佳缓解率为89.7%,15 mg BID队列最佳缓解率为80.0%。根据中期的药代动力学/药效学数据分析(发表于2023年EHA),确定RP2D剂量为10 mg BID。
--罗伐昔替尼起效迅速,81.6%(31/38)的缓解患者在4周时即达到缓解。所有患者的中位缓解持续时间(DOR)尚未达到。中位无失败生存期(mFFS)和总生存期(OS)均未达到。12个月mFFS 率为 85.2%,1年OS率为 89.6%。88.6% 的患者实现激素减量,LSS评分显示 59.1% 的患者症状有临床意义的改善。
--安全性:罗伐昔替尼在两种剂量下耐受性良好,无剂量限制毒性。没有因罗伐西替尼相关的AE导致停药。16例患者经历了≥3级药物相关不良事件(AE),AE在高剂量组更为常见。最常见的血液学AE为贫血(38.6%),而≥3级贫血发生率较低,为4.6%。
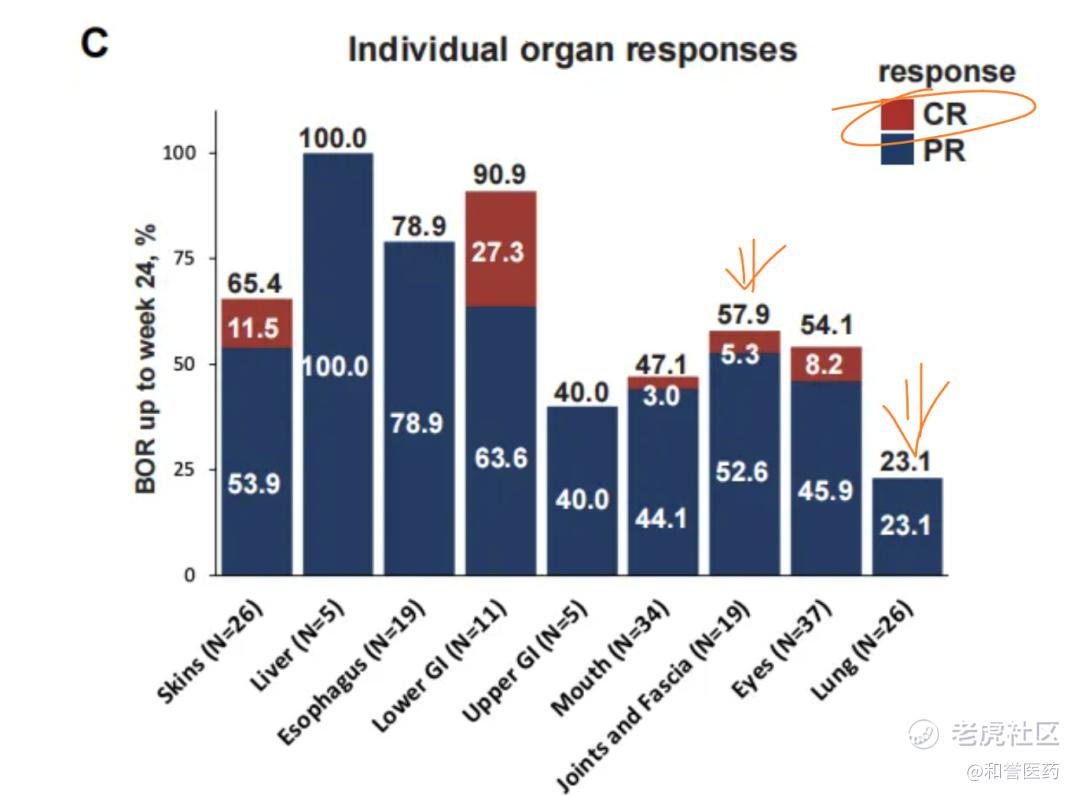
*不同部位/器官的ORR数据,关节/筋膜:57.9%(CR:5.3%)、肺部:23.1%(CR:0%)、肝脏:100%(CR:0%),数据稍逊于Incyte的CSF1R单抗。
浙大一院黄河教授/赵妍敏主任医师团队牵头的全国多中心临床研究证实罗伐昔替尼治疗激素难治慢性GVHD安全有效
https://mp.weixin.qq.com/s/6ncOP-bK2FQjDEONSpFrFQ
另外,罗伐昔替尼(TQ05105)针对中高危MF(骨髓纤维化)的适应症上市申请在2024年7月已经获得CDE受理。正大天晴于2023年美国血液学年会 (ASH) 公布了TQ05105用于治疗骨髓增殖性肿瘤 (MPN)的I期临床研究数据。结果表明,TQ05105具有良好的人体药代动力学行为,毒性可耐受,最佳缩脾率63.79%,体质症状最佳改善率为87.50%,有望为MF患者带来更多的临床选择。MF是一种弥漫性骨髓纤维组织增生性疾病,属于MPN的一种,最终会进展为骨髓衰竭或转化为急性白血病。2023年9月,原发性骨髓纤维化 (PMF) 被纳入中国《第二批罕见病目录》。目前,国内仅有芦可替尼获批用于MF患者的治疗,临床存在较大未被满足的需求。正大天晴在骨髓纤维化领域还布局了多项联合研究,如TQ05105联合BET抑制剂或BCL-2抑制剂,用于治疗中高危骨髓纤维化的临床研究,初步结果较为积极。(注:骨髓增殖性肿瘤(MPN)是一组异质性克隆增生的造血干细胞疾病,以骨髓中一系或多系增殖为特征。经典Ph阴性骨髓增殖性肿瘤包括原发性血小板增多症(ET)、真性红细胞增多症(PV)和骨髓纤维化(MF)。)

研发中的cGVHD药物:Pimicotinib/和誉
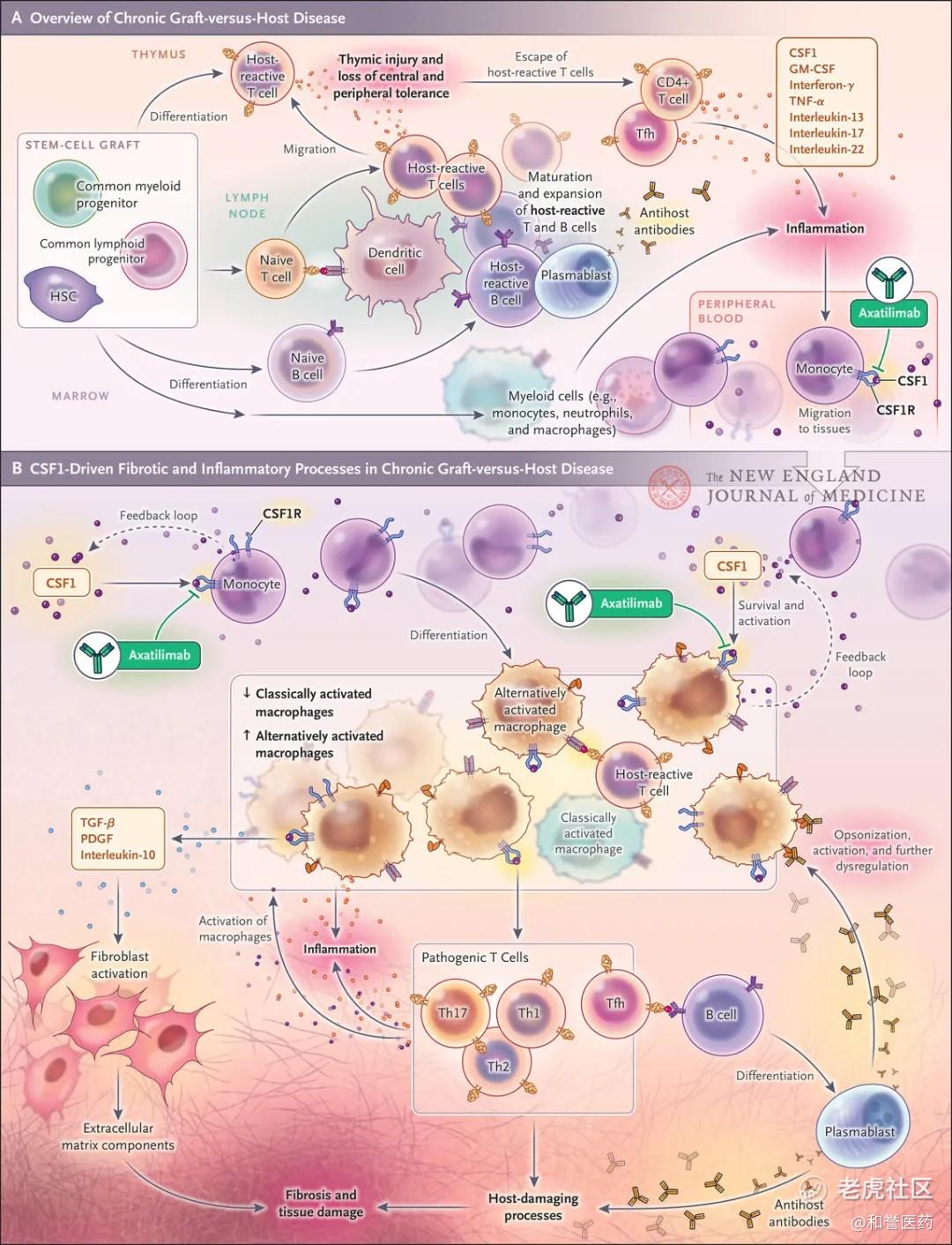
Pimicotinib和Axatilimab都是靶向CSF1R治疗cGVHD的药物,上文曾经提及CSF1R通路的一些作用方式,而cGVHD是全身性累及多个器官的免疫疾病。由于CSF1R在髓系细胞中的重要调节特性及其在骨髓、脾、结肠、肺、肝和皮肤中的广泛分布,目前研究上来看,其在各种炎症性疾病的发生和发展中起着核心作用,包括呼吸系统疾病、神经变性、肝脏炎症、动脉粥样硬化、狼疮性肾炎、类风湿关节炎和银屑病,这令到CSF1R抑制剂治疗cGVHD成为了一个研发方向。
和TGCT类似,巨噬细胞的“过度”活化释放炎症因子,慢性GVHD早期的主要特征是T细胞和巨噬细胞分泌大量的炎症细胞因子。巨噬细胞在维持慢性GVHD的持续炎症环境中发挥了关键作用,它们通过激活成纤维细胞间接导致组织损伤和纤维化。成纤维细胞则合成并分泌导致器官纤维化的细胞外基质分子。此外,巨噬细胞还产生活性氧和一氧化氮,加重氧化应激和组织损伤,并释放基质金属蛋白酶,降解某些细胞外基质蛋白,从而加剧难治性慢性GVHD中的纤维化改变。巨噬细胞还通过刺激T细胞和B细胞,导致致病性T细胞扩增和B细胞活化,进而产生抗体,可能引发慢性GVHD中的自身免疫反应。
Mohamad Mohty, M.D., Ph.D.,CSF1R Blockade for Refractory Chronic Graft-versus-Host Disease
异基因造血干细胞移植后,慢性移植物抗宿主病(cGvHD)是导致其晚期并发症和死亡的主要原因。CSF-1R驱动的巨噬细胞信号传导在炎症和纤维化发病中起关键作用,而纤维化是慢性移植物抗宿主病重要的病理特征。因此,阻断该信号传导可能有助治疗慢性移植物抗宿主病。
匹米替尼(Pimicotinib)是一种口服、高选择性且高效的小分子CSF-1R抑制剂,已在巨噬细胞驱动的腱鞘巨细胞瘤中显示出疗效,也可能具有潜在治疗慢性移植物抗宿主病的能力。本文报告了首项评估口服CSF-1R抑制剂匹米替尼在慢性移植物抗宿主病治疗中应用的初步结果(NCT06186804)。
以下是关于临床结果的数据:
--这是一项II期、开放标签、多中心研究,旨在评估匹米替尼在经≥1线的前期治疗失败的成人慢性移植物抗宿主病患者中的安全性和有效性。
--本研究分为两部分:A部分包括剂量递增和补充队列,旨在确定推荐剂量(RDs),B部分将在推荐剂量下治疗更多患者。所有患者均接受匹米替尼QD(每日一次),以28天为一个周期重复给药。主要有效性终点是根据2014年美国国立卫生研究院(NIH)共识标准定义的6个月内的客观缓解率(ORR)。次要终点包括安全性、缓解持续时间(DOR)、至缓解时间(TTR)、器官缓解率、Lee氏症状量表(LSS)评分、无治疗失败生存期(FFS)、药代动力学(PK)和药效学(PD)。
--截至2024年11月22日,共入组47例患者。在A部分,评估了4个剂量水平,包括5mg(n=1)、10mg(n=3)、20mg(n=13,包括9例来自补充队列的患者)和25mg(n=3)。未观察到剂量限制性毒性,因此选择20mg作为推荐剂量,之后继续入组了27例患者,合计40名患者。
--在可评估疗效的患者中,所有剂量水平的ORR情况为(5mg:1/1,100%;10mg:2/3,67%;20mg:25/39, 64%;25mg:1/3,33.3%)均观察到治疗反应,整体ORR达63%。在20mg在20mg QD剂量组,中位反应时间为4周。
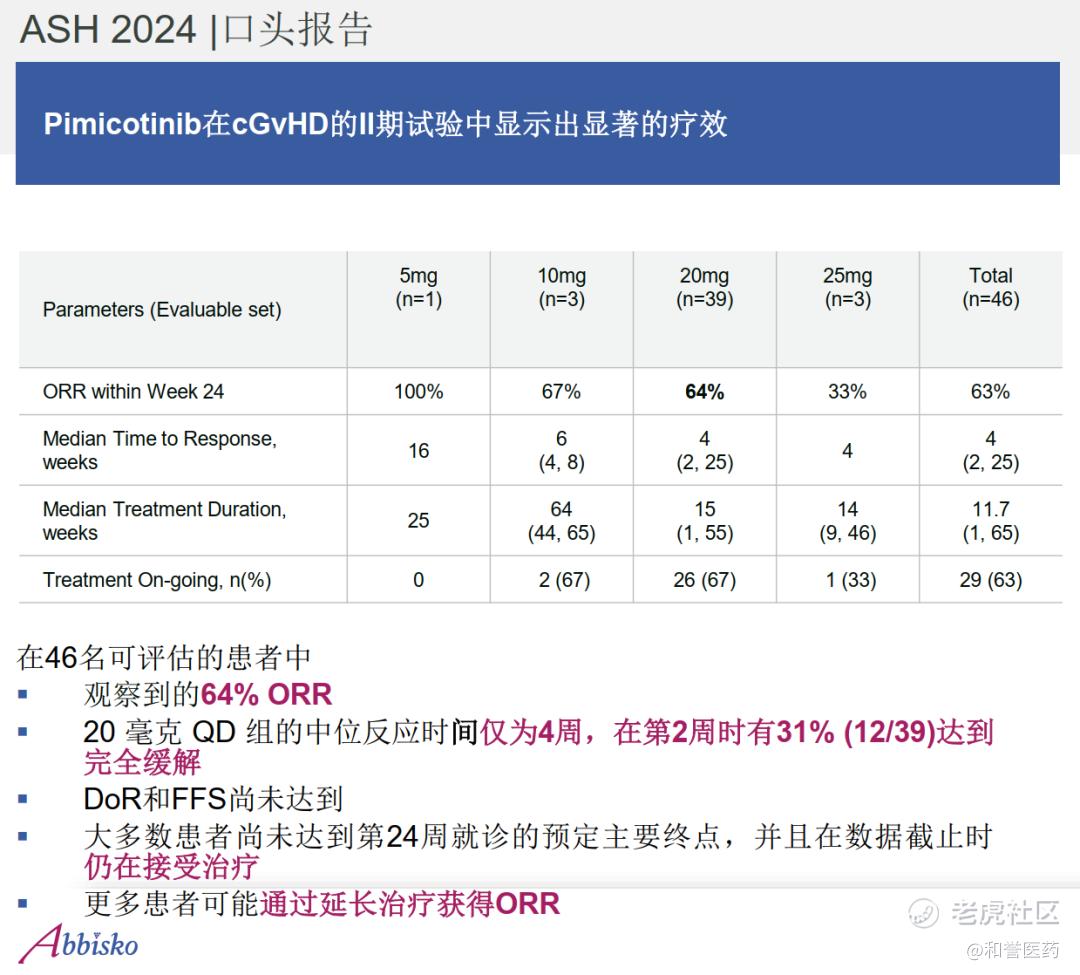
--患者中位年龄为35岁(范围:20-63岁),60%为男性。从慢性移植物抗宿主病诊断到入组的中位时间为27个月(范围:1.4-140.6个月)。57%的患者患有重度慢性移植物抗宿主病。患者接受过中位3线的前期系统性治疗(范围:1-9种),包括芦可替尼(72%,n=34)、伊布替尼(6%,n=3)和贝舒地尔(2%,n=1);基线时中位受累器官数为4个(范围:1-7个)。在数据截止时,中位治疗持续时间为12周(范围:1-65周),63%的患者仍在接受治疗。
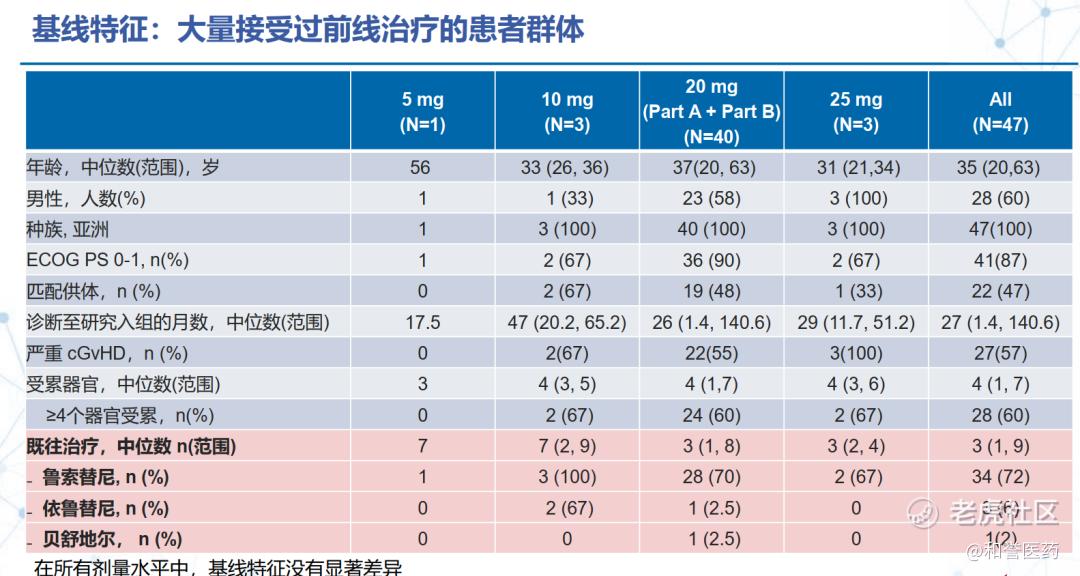
*上述基线数据已经更新了后面增补入组的20mg组的27人
--在20mg剂量下,不仅在炎症为主的器官(下消化道:50%、 上消化道40%、口腔:33%、眼睛:24%、肝脏:15%)中观察到治疗反应,还在纤维化为主的器官(包括关节/筋膜50%、食道:50%、皮肤:23%,其中1例硬皮病患者获得缓解)中也观察到治疗反应。
--匹米替尼耐受性良好,最常见的TRAEs(≥10%)包括天冬氨酸氨基转移酶(AST)升高(32%)、乳酸脱氢酶(LDH)升高(21%),丙氨酸氨基转移酶(ALT)升高(21%)、γ-谷氨酰转移酶(GGT)升高(17%),肌酸激酶(CK)升高(17%)和碱性磷酸酶(ALP)升高。这几种血清酶学水平升高,主要为1级,这些升高或与匹米替尼抑制CSF-1R后减少了巨噬细胞和肝脏枯夫(Kuffer)细胞对上述酶学的清除有关,而非引起肝脏或其他终末器官的直接损伤。
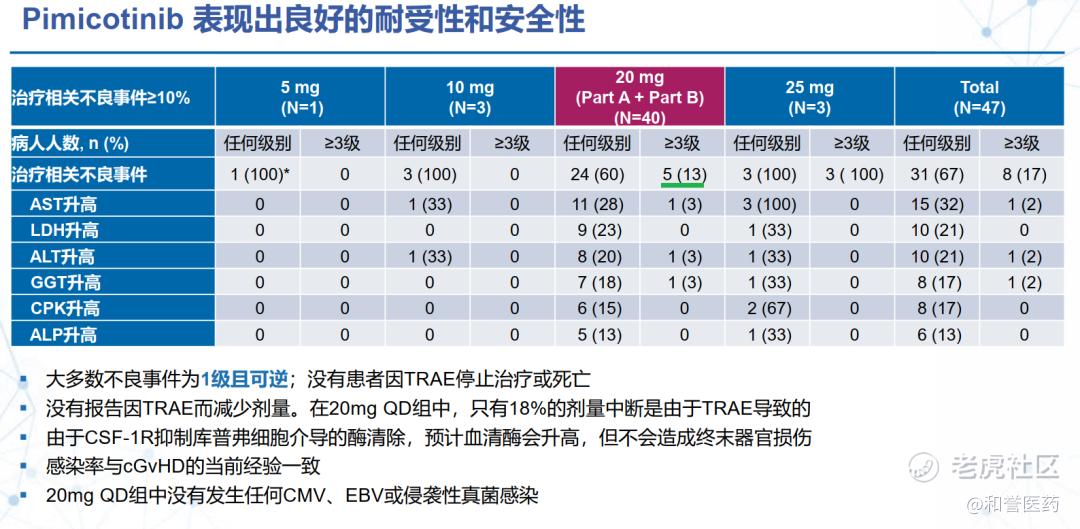
*上述安全性数据已经更新了后面增补入组的20mg组的27人
--相对于CSF1R单抗,小分子有口服的依从性优势,且在早期数据观察到和单抗相近的ORR疗效数据(64% vs 75% 6个月数据,day 1 at cycle7),以及更好的安全性数据(Grade3 TRAE 13% vs 49%),有望进一步的临床验证。

总结
针对这两个疾病做了相关整理之后,发现在不同适应症、不同靶点之间,以及药企与药企之间的映射居然如此相同。不由感叹一句……好的机会,大家都知道,好的药物开发逻辑,大家都知道,好的公司,居然也都这么相似。
而在biotech前赴后继的研发过程中,MNC继续扮演最后的护花使者角色,比如小野24亿美元收购Deciphera;Incyte与诺华(Novartis)CSO的合作;赛诺菲19亿美元收购Kadmon Holdings;以及最近Merk默克和和誉的合作……而在这些CSF1R、疾病之中,不得不佩服只有和誉把同一个药物、两个适应症都推进到即将商业化的程度,所谓“君子,和而不同”,正因为有够硬的研发能力,才能立于不败之地,既然交易金额过低的遗憾已成过去,不妨让我们期待和誉的下一个1 bn的药物。
免责声明:上述内容仅代表发帖人个人观点,不构成本平台的任何投资建议。
