低内毒素回收(Low Endotoxin Recovery,简称LER),又称为内毒素掩蔽,是指无法使用细菌内毒素检查法正常检测到样品(特别是蛋白类生物制品)中的加标内毒素的现象。制药过程中可以通过很多工艺去除微生物,但经验证能去除内毒素的工艺并不多。内毒素的监控依赖于控制和检测,如果产品有低内毒素回收现象,导致检测方法不能检测到产品中真正的内毒素水平,则产品的安全性会受到很大挑战。
本研究采用Charles River(查士利华)的市售LER缓解试剂,对部分种类的生物制品进行LER缓解策略的探索,确认生物制品内毒素检测结果的准确性。


Part 1
研究发展
LER现象由Joseph Chen和Anders Vinther在2013年佛罗里达奥兰多举行的PDA年会上首次提出,同年FDA开始要求提交生物制品许可申请(BLA)的申请人提供LER研究的相关材料。经历了对研究案例的长期收集与分析,PDA成立LER特别任务组并在2019年发布了第82号技术报告《低内毒素回收》[1],为制药公司在LER方面的研究提供了技术指导。
2025年,《中国药典》修订《9251 细菌内毒素检查法应用指导原则》[2],增加了对低内毒素回收的定义描述及缓解LER现象的方法介绍。在缓解策略方面,指导原则确认其措施应结合产品本身特性,采用在样品中添加分散剂、过量的二价金属离子、使用有机溶剂增加样品的疏水性等方式。
FDA等欧美监管机构及药典指导原则中均要求,如无法缓解低内毒素回收现象,应采用热原检查法或其他替代方法[1-2]。
Part 2
形成机制
LER可能的形成机制是基于内毒素的超分子状态发生了一种使其不易被检测到的改变。不同的内毒素对于LER的敏感性不同,这可能是内毒素脂多糖(LPS)的特异性导致的。目前已知的是由大肠杆菌制成的CSE和RSE对LER具有高度敏感性,非大肠杆菌制品或其他培养条件下的内毒素制品对LER敏感度不高。
研究表明,对于生物制品(尤其是蛋白类)样品,当配方体系中同时存在螯合剂和表面活性剂时, LER可能是一个遵循两步机制的动力学控制过程,即两步反应模型(Two-Step Reaction Model) [1]。近些年也有一些新的形成机制假说,如三步反应 “Kaz’s 模型”等[3],进一步拆解了反应过程,为LER缓解的研究提供更多思路。
除此之外,LER的发生还受到很多内外因素的影响,如时间、温度和样品处理方式等外在因素,以及配方组分、蛋白质及内毒素本身的结构等内在因素。比如含有螯合剂和表面活性剂的缓冲体系已被证明会影响LER动力学(即两步反应模型),但螯合剂和表面活性剂的种类也会很大程度上影响LER发生的概率和速度,不同的脂多糖结构也呈现出很大的差异[1]。
Part 3
研究方法
在开展LER正式研究前,一般会先进行两个步骤:LER保持时间研究预实验及缓解措施研究。

步骤1:LER保持时间研究(预实验)
01
选择一批样品进行预实验(正式LER研究时需要完成3个批次的研究),其检测方法、制备过程、数据处理方法及试剂供应商均应与日常检测一致。
02
评估产品的工艺条件,包括但不限于工艺时长、工艺温度、蛋白质、螯合剂和表面活性剂的参与等。
03
获得适宜的研究时长和研究温度,其中正式研究选择的研究时间点应不少于4个,并在相应的考察节点进行检测,预实验可根据回收率情况进行实时调整。
在评估的基础上,对未经稀释的样品原液进行加标,其加标浓度应考虑所需的稀释倍数及标准曲线中点的浓度,确认稀释后理论浓度在标曲检测范围内,以标曲中点附近浓度为宜。
若考察的节点中,将每个时间点加标样品组检测结果与加标水对照组检测结果对比,加标样品组检测回收率没有出现连续两个点低于50%,则判定样品不存在LER,可以结束预实验。若连续两个数据点低于50%的回收率则证明样品存在LER,则需展开缓解措施的研究。如果是最后一个数据点出现低于50%,需要测试额外数据点来帮助判断遮蔽现象,确认LER结果准确无误。
如果预实验结果无明显的LER,可以直接开展正式的LER保持时间的研究。如在预实验中出现了LER现象,完成缓解措施的研究后,依据研究结果,再开展正式的LER保持时间的研究。

步骤2:缓解措施研究
制药企业在进行LER研究时,产品的生产工艺往往已经确定,很难通过调整生产工艺中螯合剂、表面活性剂或其他相关缓冲液的方法来解决LER现象。因此,缓解LER主要通过调整实验室检测步骤来实现。
常见缓解LER措施包括调整样品处理方式如使用分散剂、钙离子缓冲液、镁离子缓冲液、特殊工艺的CSE或对样品进行离心、热处理等。LER缓解措施的选择会影响产品的细菌内毒素检测方法及日常放行,出于合规性考虑,从市场认可度较高的供应商处获得适宜的市售辅助试剂是更优选择。
对于存在LER现象的生物制品,缓解LER现象的方法是至关重要的。东曜平台通过与Charles River(查士利华)合作,使用Charles River(查士利华)的市售试剂对部分生物制品进行了LER缓解措施的研究。
Part 4
东曜药业案例分享(节选)

案例一 东曜抗体LER缓解策略开发与实践
针对某抗体产品,东曜在原检测方法基础上,采用多种市售试剂探索低内毒素回收(LER)缓解策略,并成功缓解了LER。
该抗体产品前期生产工艺中未添加EDTA、枸橼酸盐等较易发生LER的螯合剂,仅在原液制备段加入表面活性剂,中间产品发生LER现象的概率较小。
在正式研究开始前,对现有检测方法进行确认,选择动态浊度法进行试验。其研究结果如下表一所示:
表一 某抗体产品LER研究结果(E120加标,BET水稀释)
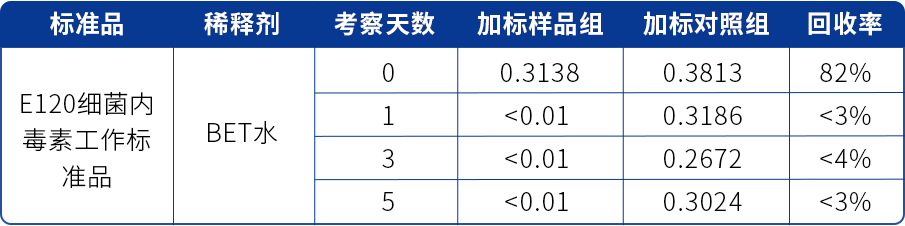
由表一可看出,该产品经E120细菌内毒素工作标准品加标,BET水稀释检测,加标第1天,加标样品组中内毒素含量已不可检出,而相同加标天数的对照组数据正常,确认存在LER现象,现有加标及试验方法需要进行调整。
从原液制备阶段开始评估产品的工艺时长及工艺温度,确认研究条件为10天,研究温度为20-25℃,在实验过程中设置4个时间点(第0, 1, 4, 10天)对产品进行LER缓解方法摸索。
首先考虑调整样品稀释剂类型,在不调整其他条件的前提下,分别使用分散剂、钙离子缓冲液代替BET水对加标样品进行稀释,其试验结果如下表二所示:
表二 某抗体产品LER研究结果(E120加标,分散剂、钙离子缓冲液稀释)
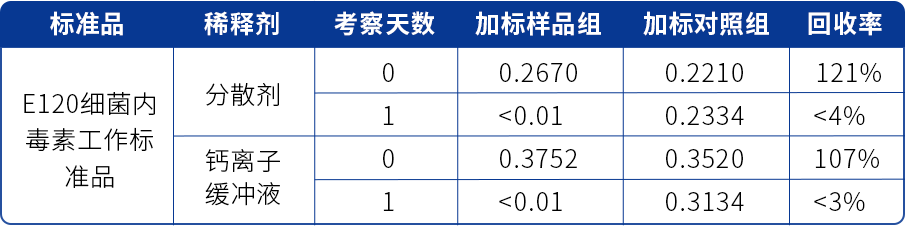
注:在缓解措施的研究中,如有需要可以尝试不同用浓度的分散剂与金属离子缓冲液进行稀释。
由表二可看出,该产品经E120细菌内毒素工作标准品加标,分别使用除BET水之外的两种稀释剂进行检测,加标第1天,加标样品组中内毒素含量已不可检出,各稀释剂相同加标天数的对照组数据正常,确认LER现象未被缓解。
经与Charles River(查士利华)技术专家沟通商讨,东曜LER研究小组引入E170细菌内毒素工作标准品,代替E120标准品对样品进行加标处理。在不调整其他条件的前提下,分别使用BET水、分散剂以及钙离子缓冲液对使用E170加标的样品进行稀释,其试验结果如下表三所示:
表三 某抗体产品LER研究结果(E170加标,BET水、分散剂、钙离子缓冲液稀释)

由表三可看出,该产品经E170细菌内毒素工作标准品加标,分别使用三种稀释剂进行检测,加标10天内其相对于加标对照组的回收率均无明显下降,判定不存在LER现象。其中分散剂稀释后加标回收率整体偏低,最终选择成分更简单、数据更稳定的BET水作为稀释剂对该产品进行正式的LER研究。

案例二 LER研究前移:以中间产品评估化解成品研究难题
成品LER研究的考察天数受产品生产工艺及周期影响,考察天数过长对于加标内毒素的稳定性也是一种挑战。对中间产品进行LER现象的研究及评估,有助于分解成品LER考察时长。对于发生LER现象概率较大的中间产品,在成品正式研究前进行确认,缓解其LER现象。
某抗体产品由于工艺需要,在生产过程中多次加入EDTA、枸橼酸盐及吐温等金属螯合剂和表面活性剂,因此中间产品存在LER现象的概率较高。
根据评估结果,对该抗体产品的某中间产品进行LER研究,在正式研究开始前,对现有检测方法进行确认,选择动态浊度法进行试验。其研究结果如下表四所示:
表四 某抗体中间产品LER研究结果(E120加标,BET水稀释)
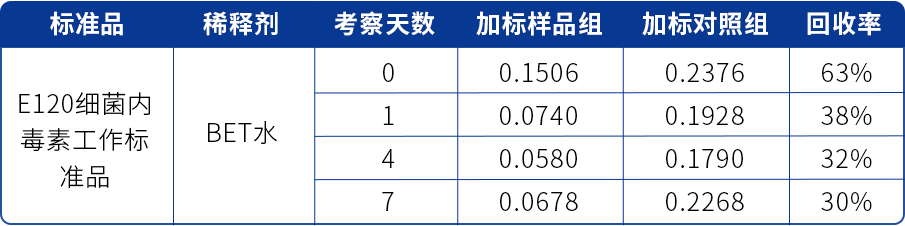
由表四可看到,该产品经E120细菌内毒素工作标准品加标,BET水稀释检测,加标第1天、第4天,加标样品回收率分别为38%、32%,连续两个时间点回收率<50%,判定样品存在LER现象。
引入E170细菌内毒素工作标准品,分别使用分散剂及镁离子缓冲液代替BET水对加标样品进行稀释,其试验结果如下表五所示:
表五 某抗体中间产品LER研究结果(E170加标,分散剂、镁离子缓冲液稀释)
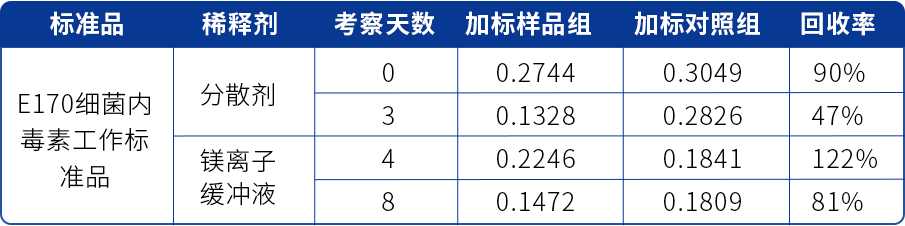
由表五可看出,该产品经E170细菌内毒素工作标准品加标,使用分散剂作为稀释剂进行检测,加标第3天,加标样品回收率已不满足要求,随即在第4天、第8天改换镁离子缓冲液作为稀释剂进行检测,第8天加标样品回收率为81%,满足LER研究要求,最终选择符合条件的镁离子缓冲液作为稀释剂,进行正式的LER研究。
Part 5
总 结
生物制品LER的发生机制相对复杂,不易被预测,因此这项研究需要很长的试验周期和投入大量的资源。而缓解产品LER现象的过程更极具挑战性。LER研究推进的顺利程度与否与产品本身的特性、研究团队对产品的了解以及研究团队缓解LER现象的经验等有很大关联。
除此之外,如果试剂供应商无法提供多种缓解LER现象的试剂选择,产品LER现象无法缓解,不得不在中途变更鲎试剂厂家或检测方法,将严重耽误药品的申报进程。基于合规性及研究可行性角度考量,在前期调研内毒素检测试剂供应商时,应优先选择拥有配套试剂品类齐全、供货稳定、市场认可度高的供应商。
无论是从美国FDA的BLA提报要求,还是从2025版中国药典9251章节新增LER内容,都不难看出,LER研究已经成为生物制品上市申报过程中的必经之路。
结合东曜目前的项目经验,在2025版本药典发布后,中国监管在生物制品上市申报时,会要求进行LER研究。
东曜药业在LER研究具有多年的丰富经验,目前已建有生物制品LER研究及分析平台,基于客户CDMO项目的实际需求,提供LER研究及解决方案,保证产品检测结果的有效性,满足产品上市申报的需求,保障临床用药安全。

点击下方“阅读原文”,获取PDF文档。
参考资料
[1]
PDA Technical Report No.82 Low Endotoxin Recovery.
[2]
中华人民共和国药典:2025年版 四部[S]. 北京:中国医药科技出版社,2025: 9251.
[3]
Mechanism of Low Endotoxin Recovery Caused by a Solution Containing aChelating Agent and a Detergent, Immunome Research, Vol.15 Iss.1 No:166
关于东曜药业
About Us
东曜药业(股票代码:1875.HK)一家全球领先的生物药合同研发生产组织(CDMO),致力于成为行业领先、客户信赖的生物医药最佳合作伙伴。依托先进的一站式产业化平台,我们提供以抗体为代表的蛋白类药物、生物类似药,以及以ADC为代表的偶联类药物从早期研发到商业化生产的全流程CDMO服务,加速客户项目进程。
东曜拥有符合GMP规范的大规模生物药商业化生产基地,以及符合中/美/欧标准的国际化质量管理体系。凭借先进的技术平台和专业的技术及服务团队,赋能海内外不同地区与国家的客户项目,业务覆盖欧美、亚洲及众多新兴市场。
东曜药业秉承“以品质、助创新、共成长” 的服务理念,结合经验丰富的团队,依托创新内核,坚守合规和质量基石,构建生态协同发展路径,赋能合作伙伴,共创双赢。
了解更多,请访问:www.biodlink.com
精彩评论